
未依國際標準制定,國內化粧品GMP未順利接軌遭國際拒絕
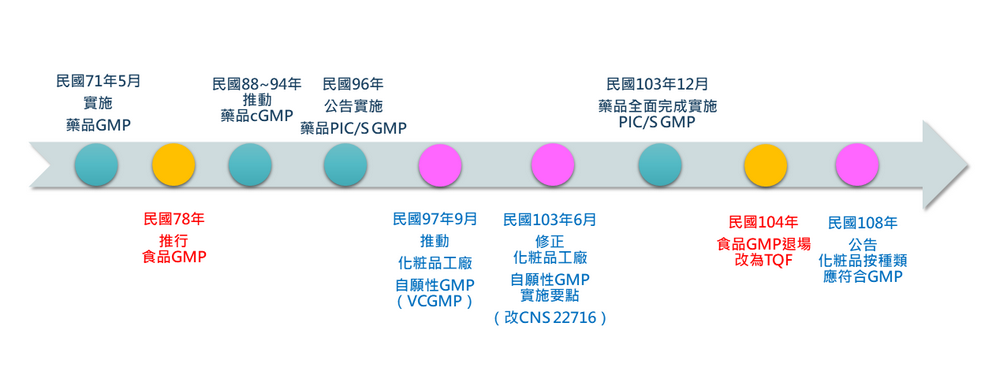
我國GMP目前發展最晚的是化粧品產業,民國97年由當時已改制的食藥署與經濟部工業局,共同推動化粧品工廠自願性GMP認證機制,也就是俗稱的VCGMP(Voluntary Cosmetic Good Manufacturing Practice)。截至民國102年底,全台有86家申請查核、55家通過,申請通過驗證之品項包含化粧水、乳液、精華液、洗髮精、沐浴乳、面膜、口紅、粉餅、眼影等共計644種。整體通過率大約六成多,取得難度高於業界所期待。
然而,推動自願性化粧品GMP時在外銷市場上出現一困境,當年自願性GMP規範內容未引用ISO 22716,以致產品外銷時,國外客戶對此產生疑慮。為解決此一困境,民國103年時,主責的兩個單位-衛福部與經濟部,公告修正「自願性化粧品優良製造規範(GMP)實施要點」,並以我國國家標準-CNS 22716作為驗證品質管理系統之依據,藉此接軌國際標準規範ISO 22716。截至111年1月,通過自願性GMP的業者共有64家。
 圖五、台灣藥品、食品、化粧品GMP實施進程
圖五、台灣藥品、食品、化粧品GMP實施進程
從發展沿革中,更細部向大家說明國外市場為什麼對最初的自願性GMP感到疑慮。在推行自願性化粧品優良製造作業規範,並未引用ISO 22716,因此在部分章節會略有差異,證書上也不會出現ISO 22716的字眼。對於國外市場而言,就很有可能被視為不一樣的規範而不被認同。
經濟部與衛福部期盼能夠讓業者有更高的動機進行申請,針對第一版的自願性GMP進行調整。最關鍵的就是改變驗證依據,將國際標準轉為國家標準,在民國101年進行公告、 民國102年進行相關修訂,將國家標準中的章節與國際標準一致化。
ISO 22716是由國際標準組織(ISO)所製定出來的,它與ISO 9001品質管理系統並不相同,ISO 22716是特定用於化粧品行業的實務管理規範。在歐盟與東協的管理規定中皆提出化粧品的製造要符合ISO 22716,因此國內業者若要外銷的情況下,勢必要遵守相關規範。
化粧品優良製造準則於民國108年8月13日公告以後,在第一條即提到此製造準則是依據國際標準組織化粧品優良製造規範ISO 22716來制定。修正後的新法僅在第十七章新增了附則,至於中間章節其實與國際規範相似。
實務上來說,許多人分不清楚ISO 22716、自願性GMP或是CNS 22716,這邊幫大家進行總結。VCGMP就是國內常稱的自願性GMP,其採用的是CNS 22716,而CNS 22716是依循ISO 22716的條文內容制定。因此,在在文章架構與要求上,其實三種規範是幾乎相同,沒有太大的差別。更簡單來說,新法所推行的化粧品優良製造準則,其實就是將ISO 22716納入國內行政法規內。
 圖六、我國已將國際規範納入《化粧品優良製造準則》
圖六、我國已將國際規範納入《化粧品優良製造準則》
在實際執行驗證的情況中,常見國內業者其實對相關法規的瞭解程度並不高,甚至可能出現誤解或迷思,進而導致採取的方式較為耗費資源,甚至是選擇錯誤的方式因應,而造成資源的浪費,實屬可惜。若能對於法規更加瞭解,就能越清楚法規的需求。也就能更有效運用資源,將資源花在刀口上。
根本上的差異導致藥品、食品、化粧品業者命運大不同
從藥品GMP的角度來看化粧品法規,兩者有根本上的差異。依據藥事法第57條第2項有提到,藥物的製造它的廠房、設施、設備、人事、生產、品質管制、運輸等,都必須符合藥物優良製造準則的規定,且須經過中央衛生主管機關檢查合格,並取得藥物製造許可後,才能進行製造。
若上述任一條件沒有符合就是製造偽藥,屬於違法行為。因此藥廠是嚴格控管的,不僅僅只要符合法規,還需要經主管機關檢查合格,才能開始生產。這也是為什麼在實施藥品GMP的過程中,藥廠相對減少的原因。對於業者而言,在檢查不合格的情況下,只能選擇加碼投資,以達到檢查合格,或者選擇結束營業。
然而,依據化粧品衛生安全管理法第8條的2項,經中央主管機關公告之化粧品種類其化粧品製造場所應符合化粧品優良製造準則,中央主管機關得執行現場檢查。雖然,法規賦予中央主管機關到現場查核的權責,但並未提到必須要檢查合格才能製造。相對來看,僅要求業者應該要符合法規,這是與藥品GMP不同的地方。因此,預期新法雖然會對化粧品產業造成影響,但不會出現如藥廠一樣大量減少的現象。
先前提到,法規賦予中央主管機關到現場查核的權責,在新法第13條第一項中也提到,主管機關可以派人查驗國內業者,抽查內容包括設施、PIF、產品供應來源等紀錄資料,或針對使用的原料進行抽驗、檢驗。在此也要提醒相關業者務必配合,不得規避、妨礙或拒絕。
同時,我們也可以將化粧品GMP與食品GMP進行比較,因為這兩個規範與藥品GMP的主管機關皆相同。將化粧品、食品及藥品GMP三者並列比較,可以看見與化粧品GMP相同的是,食品GMP法規中並未要求食品業者必須要經主管機關檢查合格才能進行製造。這其中的差異便是,化粧品、食品及藥品,三者預期的消費對像不相同。因此,在管理上的嚴謹度會有相當大的差距。以食品與化粧品來看,消費的對象主要來自一般民眾為主。至於藥品,消費族群主要是病人,因為一般大眾在身體健康正常的情況下,並不會去服用藥物,僅會為了達成某些醫療目的才會使用。
雖然化粧品法規相對較為寬鬆,但是業者仍應積極準備,由於主管機關可以執行現場檢查,未來可以預期的狀況是,主管機關可能藉由推動或啟動專案,針對特定產品類型的相關業者進行稽查。以食品業為例,即依照風險程度高低進行抽查,進而產生不同的稽查專案,譬如:逢年過節針對中式炊蒸食品的製造業者進行專案稽查,以確保民眾食用安全。
雖然化粧品GMP存在不須主管機關檢查合格,也可以製造的情形下,也不代表業者可以放鬆。依據化粧品衛生安全管理法第16條中提到,若製造工廠未符合設廠標準或未符合GMP的情況下,經主管機關認定有害衛生安全,主管機關可以要求不得供應、販賣、贈送、公開陳列甚至不能供人試用。第17條同時規定,經認定有害衛生安全,可以要求業者回收產品。甚至在第18條中可以要求將違法的產品沒入銷毀。經令限時改正,屆期不改正,則會依第22條規定,可處新臺幣2萬元以上500萬以下的罰鍰,並得按次處罰,情節重大者,得要求停業,甚至廢止業者的登錄、許可證或工廠登記等等。由此可看,主管機關設立新法的目的並不是希望業者因新法而停業而是期待業者善盡管理責任,在一般情況下要求業者限期改正,除非惡意不改或有害衛生安全情節重大,才會令其歇業、廢止其部分登記等。
對相關法規有初步的瞭解後,接下來要瞭解該如何因應。首先,要確定工廠自身的合法性,新規中化粧品工廠具有設廠標準,除非是做手工皂或相對小型的業者,不用辦工廠登記。若已經達到需辦理工廠登記的標準,即便是手工皂業者,仍需辦理工廠登記。
如不能肯定是否需進行工廠登記,需依各縣市工廠管理機關(如:經發局)規定進行辦理。另外,因為我國工廠相對較小,或是工廠設立年代較久遠,其所處位置並非現今的工業用地。業者可依據工廠管理輔導法,申請納管證明。但要注意的是納管證明具有期限,因此業者需確認納管期限的合法性。
在導入化粧品GMP的過程中,因需符合管理制度及軟硬體的部分,業者需檢視軟硬體是否需要更新或建置。在新法中並未規定要如何符合法規,僅由業者依據自身的條件選擇。對業者來說,可以選擇的方式包括自我宣告、尋找第三方驗證機構,或是參加自願性GMP等方式。以監管的角度來看,不管業者選擇哪一個方式,主管單位都還是可以執行現場檢查、產品抽驗等。差別僅在於若業者並非取得自願性GMP或第三方驗證, 可能會被主管機關列為優先稽查的對象,因為站在主管機關的角度來看,業者自我宣告的監管風險較高。
 圖七、ISO 22716 :2007驗證流程
圖七、ISO 22716 :2007驗證流程
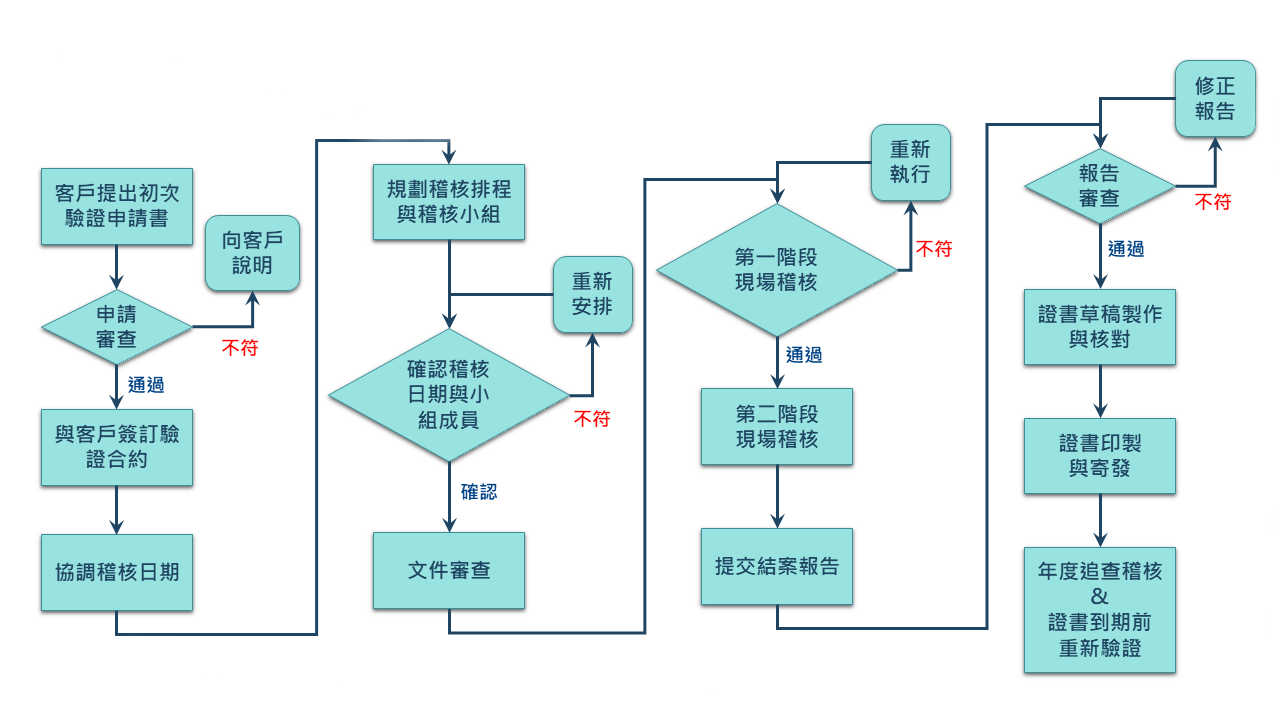
業者在進行選擇時,最關鍵的是要依據自身需求,避免因自身資源限製而提高不符合GMP的情況,業者若不想採取自我宣告,希望能採取積極的方式,首先可以選擇第三方驗證。由於查驗機構不同,與自願性GMP嚴苛的通過率相比,由於,第三方驗證的通過率相較來得高,是較彈性且務實的。
不論是自願性GMP或是第三方驗證,對業者而言都是一筆費用的支出。第三方驗證與自願性GMP的證書效期都是三年。所以業者如何選擇查驗機構好讓證書效益發揮到最大,才能讓每分錢花在刀口上。身為全球最大化粧品集團所在國-法國的法國標準協會(Afnor),依循著ISO 17021等認證管理規範運作,驗證流程嚴謹,不僅會安排年度的稽核,每年也會執行現場稽核,確保業者維持良好的管理,降低違法風險,贏得其客戶的信賴與持續交易的信心。
1. 是否一定要Class 10萬的無塵室才能取得GMP?
實務上常聽到是否一定要Class 10萬等級的無塵室,或是相關硬體等級是否需要非常高才能取得GMP。法規上來說,ISO 22716並沒有提到一定需要無塵室,只要滿足GMP的基本需求,例如:地板、牆壁、天花板之樣式或材質,不要造成廠區汙染即可。當然若業者有設無塵室,對於查核人員來說,符合法規的信心會更高。
依照目前我國化粧品產業的風氣,國內一千多家業者中,過半是屬於代工業者。代工廠的目標是希望爭取大量代工訂單,因此若廠內有相對較優質的條件,也比較容易獲得訂單,當然這也會在業界形成競爭優勢。因此,無塵室是個選項,但並不是絕對的選項。
2.是否一定要品管人員?
在實務中也經常有人詢問此問題,根據ISO的內容中有提到品質的權責人員,因此預期廠區需要有品管人員,而這個品管人員通常會獨立於在生產端。若能在GMP文件出示相關紀錄,在實務上來說,其實是有較高的優勢。一般會建議業者,需要有專人來負責品管,因為從原物料、物料、半成品、成品到後續生產紀錄、品質檢驗的紀錄等文件皆需要品管人員。所以對業者而言,導入GMP在軟體上是需要付出資源的。
3.是否一定要有品管實驗室?
國內有些業者因為規模並沒有到很大,設置品管實驗室可能會佔據工廠部分空間,因而產生困擾。在ISO 22716的章節中,除了第九章提到要有品管實驗室以外,在第十二章分包的部分,也將檢驗納入分包的項目之一。所以實務上,如果業者有實驗室,代表工廠擁有較佳的條件,業者接下來必須依照ISO 22716中第九章的規範進行相關管理。若今天受限於工廠規模,暫時無法設置實驗室,業者可選擇透過分包的方式,來符合要求,因為依照目前的設廠標準,並沒有硬性規定此部分。
4.要準備多少文件才能驗證?
衛福部已經將化粧品製造場所實施GMP文件清單與標準作業程序相關範例放在官網上,提供給業者取用。業者可以利用這些公版文件,進行調整,依照工廠規模、流程的複雜度進行修正。此公版文件僅是提供業者在有時間壓力的狀態下,以最便捷的方式進行調整,因此並不建議業者完全照抄。若業者不確定該如何著手準備,官方至少提供了最基礎的文件,以輔導代替強制,供業者強化自我管理。
5.要準備多久的紀錄才能申請?
在決定實施驗證的情況下,也會關係到要文件的建立,究竟要準備多久的紀錄才能進行申請呢?如果業者申請自願性GMP,依照規定須要實施滿三個月,或者產品連續生產三批,才能提出申請。這部分其實沿用食品GMP的規定。早期工業局在進行食品GMP時,也有同樣的要求,其目的是希望業者的品質是具有穩定性的,因為不管是製造過程或是導入文件,廠內相關作業人員需要習慣的養成。一般來講,三個月約莫是習慣養成的時間點。至於產品至少可能連續生產三批,是因為比較能夠再現工廠的製造品質。因此,會建議業者至少達到這兩項要求再提出申請,較能提升驗證過程中的信心度。當然,若有較為特殊的情況,會以個案方式來處理。原則上第三方驗證機構,每年都會對業者進行相關的稽查。
必須要鼓勵各位,化粧品GMP需要時間的養成,一旦開始導入,就要持續的維持,累積足夠的相關紀錄,對驗證機構來說,會更具有發證的信心。
- 資料來源:台灣貝爾國際驗證機構